Why the “biology-first, modality-agnostic” era is eclipsing yesterday’s size-based dogma
From bucketed modalities to a continuum mindset
For most of the modern drug-discovery era, chemists and biologists organised therapeutics along a rigid, size-driven taxonomy: small (< 500 Da), peptide, protein, gene, cell. That reductionist view served medicinal chemistry well when Lipinski’s rule-of-five was the dominant heuristic and gate-keeper of oral success, and when antibodies, oligonucleotides and viral vectors each lived in their own, distant technological silos.
Yet 2025’s pipeline tells a different story. Modalities have escaped their buckets, merging form factors, and combining mechanisms of action into a continuous drug design real estate spanning six orders of magnitude from sub-100 Da molecular glues to 10^8 Da cell products. The locus of decision-making has therefore inverted: discovery programmes now start with the biology—the molecular aetiology of the disease, the spatiotemporal context of the target, the desired pharmacodynamic half-life—and only then is a modality chosen (or engineered) to satisfy that need. Drug hunters can consider any chemistry–biotechnology hybrid from an integrated toolbox in an increasingly modular fashion, rendering the question “small or large molecule?” borderline obsolete.
"New approaches to treatment require us to bring forward innovative approaches to drug discovery."
Dr Susan Galbraith, Executive Vice President of Oncology R&D at AstraZeneca, has been a prominent voice in highlighting the transformative impact of emerging therapeutic modalities on drug discovery.
The result is a combinatorial explosion of design space and an invitation to innovation, unlocking new challenges and opportunities at the intersection of chemistry, biology, and biotechnology. Let’s examine this evolution through a (non-exhaustive!) sample of today’s therapeutic landscape.
1. Small is growing up: new TACtics and chemical matter beyond rule-of-five
1.1 bRo5, macrocycles and constrained peptides
The chemical space once thought “too big to be drug-like” is now a fertile hunting ground for ligands of protein–protein interfaces, RNA structures and membrane transporters. bRo5 macrocycles (MW 700–1400 Da) retain passive membrane permeability through conformational chameleonicity, while hydrocarbon-stapled α-helices provide protease-resistance and pre-organised binding entropy. DNA-encoded libraries that safely wander into bRo5 space are identifying hits against notoriously shallow surfaces such as KRAS-G12D and TEAD.
1.2 Proximity-based modalities, targeted protein degradationand molecular glues
Platform innovators have weaponised the cell’s own proteolysis machinery. Second-generation PROTACs such as ARV-766 show clinically meaningful PSA50 responses in metastatic castration-resistant prostate cancer without dose-limiting toxicities, confirming that catalytic degradation can overcome point-mutation resistance seen with classical occupancy inhibitors. Meanwhile, structure-guided “molecular glues” collapse the footprint to < 450 Da—below the oral sweet spot—dissolving yet another size boundary. ARV-766 Demonstrates Clinical Activity in AR LBD–Mutated ...
1.3 Ribonuclease-Targeting Chimeras (RIBOTACs)
By chemically hijacking RNase L, sub-kilodalton chimeras now achieve sequence-selective RNA degradation from a purely small-molecule scaffold, further eroding the historical wall between nucleotide therapeutics and heteroatom-rich medicinal chemistry. RIBOTAC technology conjugates RNA-binding small molecules to RNase L recruiters. The pre-miR-155 RIBOTAC reduced mature miRNA levels by 71% in lymphoma models through site-specific cleavage at 5'-UUU/3'-GUC motifs. Structural optimization of the RNase L-binding domain improved catalytic efficiency (kcat/Km = 4.2 × 10^5 M^-1s^-1) by enhancing ubiquitin-like domain engagement. Programming inactive RNA-binding small molecules into bioactive degraders | Nature
1.4 Lysosome-Targeting Chimeras (LYTACs): Beyond Proteasomal Degradation
Aptamer-LYTACs for Precision Targeting
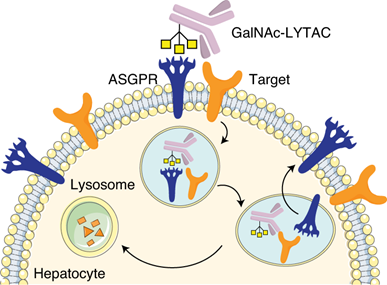
Aptamer-LYTACs represent a significant advancement over antibody-based systems, combining the specificity of nucleic acid aptamers with tri-GalNAc ligands for hepatic targeting. These chimeras (MW ~3.4 kDa) enable liver-specific degradation through asialoglycoprotein receptor (ASGPR) engagement. For instance, EGFR-targeting Apt-LYTACs reduced receptor levels by 80% in hepatocytes within 24 hours while sparing non-liver cells. The modular design allows rapid synthesis compared to antibody conjugates, with degradation efficiency depending on aptamer binding affinity (Kd < 50 nM optimal) and linker rigidity. Aptamer-LYTACs for Targeted Degradation of Extracellular and Membrane Proteins - PubMed, LYTACs that engage the asialoglycoprotein receptor for targeted protein degradation - PMC.
Nano-LYTACs for Enhanced Tissue Penetration
Self-assembling peptide-GalNAc nanospheres (15-20 nm diameter) demonstrate improved tumor penetration over conventional LYTACs. The Nanosphere-AntiCD24 system degraded >90% of CD24 in triple-negative breast cancer models within 48 hours, disrupting the CD24/Siglec-10 immune checkpoint. When combined with glucose oxidase, these nanocarriers induced synergistic oxidative stress and macrophage reactivation, reducing tumor volume by 68% in murine xenografts compared to monotherapy. Nano‐LYTACs for Degradation of Membrane Proteins and Inhibition of CD24/Siglec‐10 Signaling Pathway - PMC
Bispecific VED-LYTACs for Ocular Therapies
VED-LYTACs combine M6PR-targeting aptamers with VEGF-binding motifs through stable phosphorothioate linkers. In laser-induced choroidal neovascularisation models, intravitreal administration (2 μg/eye weekly) reduced vascular leakage by 82% through lysosomal VEGF degradation, outperforming anti-VEGF antibodies (54% reduction).The bispecific design enables simultaneous engagement of M6PR and VEGF (EC50 = 12 nM), circumventing compensatory signaling pathways in neovascular eye diseases. Targeted degradation of VEGF with bispecific aptamer-based LYTACs ameliorates pathological retinal angiogenesis - PMC
1.5 Autophagy-mediated Degradation Systems (AUTACs)
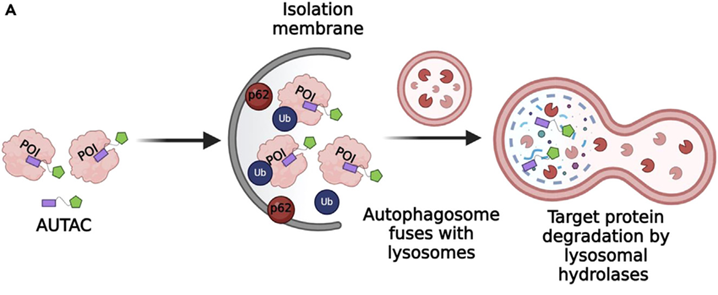
AUTACs for Organelle Clearance
AUTAC molecules utilise 8-nitrocyclic GMP tags to induce K63-polyubiquitination and subsequent mitophagy. The mitochondrial-targeting AUTAC Mito-AUTAC-1 (MW 589 Da) cleared >75% of fragmented mitochondria in Down syndrome fibroblasts within 6 hours, restoring OXPHOS capacity by 2.3-fold.Structural optimisation revealed the critical role of the para-fluorobenzyl group in enhancing lysosomal recruitment, with EC50 improving from 1.2 μM to 180 nM through halogen positioning. AUTACs: Cargo-Specific Degraders Using Selective Autophagy - PubMed
ATTECs for Lipid Droplet Removal
LD·ATTECs employ cholesterol-derived tethers to link lipid droplets to LC3-binding domains. The lead compound ATTEC-LD3 reduced hepatic triglyceride content by 64% in NAFLD models through selective autophagic degradation, outperforming FXR agonists (38% reduction).Crystallography studies revealed a unique binding mode where the cholesteryl group inserts into lipid droplets while the adamantane moiety engages LC3's hydrophobic pocket (ΔG = -9.8 kcal/mol). Beyond targeted protein degradation: LD·ATTECs clear cellular lipid droplets | Cell Research
1.6 Deubiquitinase-Targeting Chimeras (DUBTACs): Protein Stabilisation Strategies
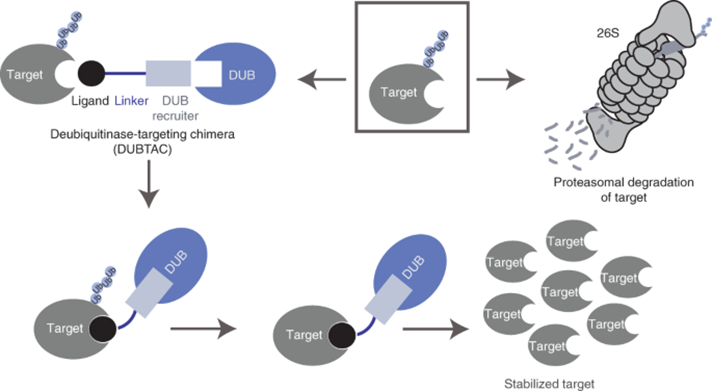
OTUB1-Based DUBTACs
The first-generation DUBTAC stabilising ΔF508-CFTR combines lumacaftor with EN523, a covalent OTUB1 binder targeting C23 allosteric cysteine. This chimera increased CFTR membrane localisation 4.2-fold in bronchial epithelial cells, restoring chloride conductance to 78% of wild-type levels. Pharmacokinetic studies showed lung-selective accumulation (tissue/plasma ratio = 8.7) due to lumacaftor's bronchoaffinity. Deubiquitinase-targeting chimeras for targeted protein stabilization | Nature Chemical Biology
USP7-DUBTACs for AMPK Stabilisation
Novel USP7-based DUBTACs stabilise AMPKβ isoforms through non-covalent recruitment. The AMPKβ1-specific DUBTAC (MW 832 Da) increased hepatic AMPK activity 3.1-fold in diabetic mice, reducing HbA1c by 1.8% through dual phosphorylation at Thr172 and Ser108. Isoform selectivity stems from varying linker lengths between the USP7 ligand (XL177A) and AMPK-binding moieties, with C8 spacers favoring β1 over β2 (8:1 selectivity). USP7-Based Deubiquitinase-Targeting Chimeras Stabilize AMPK | Journal of the American Chemical Society
Current efforts focus on developing non-covalent DUBTAC recruiters beyond OTUB1/USP7. The recent identification of USP28 ligands (IC50 = 8 nM) enables stabilisation of DNA repair proteins like 53BP1, potentially enhancing radiotherapy efficacy. Similarly, HECT-type E3 ligase recruiters (e.g., NEDD4-1) are being explored for tissue-specific degradation. USP7-Based Deubiquitinase-Targeting Chimeras Stabilize AMPK | Journal of the American Chemical Society
2. Large is getting leaner: antibody engineering on an industrial scale
2.1 Nanobodies (VHHs) offer compelling properties bridging small and large molecules
Nanobodies (15 kDa), the variable domains of camelid heavy-chain-only antibodies (VHHs), represent a paradigm shift in antibody engineering. Their distinctive features include:
- Single-domain architecture: Lacking light chains, with antigen recognition mediated solely by three extended CDR loops (CDR3 up to 24 residues vs. 12 in conventional Abs) Nanobody or VHH - What's the difference?, NANOBODIES®: A Review of Diagnostic and Therapeutic Applications
- Enhanced solubility: Hydrophilic substitutions (F37/E45/R45/G47) in FR2 region replace hydrophobic residues in conventional VH domains VHH Nanobodies: Revolutionizing Antibody Applications
- Thermal stability: Retain binding capacity at 90°C and resist aggregation at 4 mg/mL concentrations Statistical Analysis of Nanobody Sequences and Structures with Molecular Dynamics Simulation of Nanobody VHH
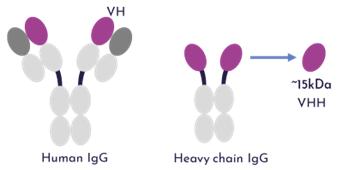
These elegant biologics are enjoying a surge in popularity and design innovation driven by computational and AI approaches, such as AlphaFold 3 and advanced protein language models. To support this surge in innovation, automation platforms such as Arctoris’ Ulysses® are manufacturing and characterising thousands of de novo proteins each month.
2.2 ADCs 2.0—chemistry meets biologics
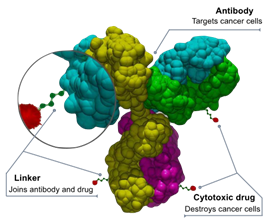
With 17 marketed ADCs and global sales exceeding $10 billion, the format has graduated from proof-of-concept to validated, programmable platform. (Global Sales of Antibody-drug Conjugates (ADCs) in 2024) The January 2025 approval of datopotamab deruxtecan (Datroway) in HR+ HER2-negative breast cancer underlines how site-specific DAR control and topoisomerase-I payloads can turn previously “chemo-refractory” histologies into ADC territory. (A New 'Missile' Drug Is Approved To Treat the Most Common Breast Cancer Type) State-of-the-art THIOMAB™ and glycoengineering now deliver sub-25 % DAR heterogeneity and millimolar serum stability, while hydrophilic self-immolative spacers mitigate off-target neutropenia.
2.3 Bispecifics, trispecifics and T-cell engagers
Bispecific engineering has shifted from bespoke antibodies to modular Lego®-like assembly. IgG-scaffolded tarlatamab (Imdelltra) earned FDA approval in 2024 as the first T-cell engager for small-cell lung cancer, validating high-potency immune redirection in solid tumours. (First-in-class T cell engager approved for lung cancer - Nature) Next-generation “MuTICs” layer a third specificity—often PD-L1 or CD47 blockade—to synchronise synapse formation with checkpoint release, narrowing the mechanistic gap between biologics and engineered cell therapy.
3. Hybrids in the middle: conjugates, nucleic acids and programmable carriers
3.1 Peptide-drug conjugates and cell-penetrating tags
Approved radioligand Lutathera proved that a 1.3 kDa octreotate peptide could chauffeur β-particles into neuroendocrine tumours. Iterations now graft cell-penetrating motifs (e.g., penetratin, TAT) or lipid handles to traverse hepatic endosomes, exemplifying how “medium” peptides borrow tricks from both small molecules and proteins.
3.2 GalNAc and oral oligonucleotide delivery
Targeted delivery of siRNA to hepatocytes via tri-antennary GalNAc set the standard for sub-cutaneous dosing; the next leap is oral nucleic-acid therapy. Enteric-coated nanoparticle formulations and transient tight-junction modulators have moved two siRNA candidates into first-in-human studies, riding a 150-trial wave that has pushed RNAi’s CAGR close to 80 % over the past four years. (Novotech Report Reveals Growth in siRNA Research: Over 150 ...)
3.3 Engineered exosomes and extracellular vesicles
Rationally loaded exosomes (< 200 nm) combine the biodistribution of liposomes with the endogenous stealth of cellular membranes. More than 120 candidates—40 % already in clinical phases—populate the 2025 exosome drug pipeline, the majority exploiting surface-displayed ligands for tissue-specific docking. (Exosome Therapeutics Market Report 2025: 120+ Drug)
Overview: The Continuum of Therapeutic Modalities
† Ranges capture the bulk of reported drug-like examples; outliers exist.
‡ Exosomes are particles, so size is conventionally reported in nanometres rather than Daltons.
Outlook—towards “composable” therapeutics
The therapeutic landscape of 2025 represents a significant shift towards modularity and a continuum of molecular modalities that transcend traditional weight-based classifications and design constraints. Therapeutics are increasingly tailored with specificity to target biology, tissue selectivity, and underlying disease mechanisms. With advances in precision medicine, catalytic degradation and stabilisation technologies, drug discovery now offers precise spatial control (e.g., organelle- or tissue-specific targeting), refined temporal regulation (such as photoactivated PROTACs), and sophisticated combinatorial logic (e.g., AND-gate degraders) that activate selectively within disease-relevant contexts.
This expanding and versatile toolkit compels us as drug discovery experts to embrace broader, integrative approaches and to continually expand the scope of our scientific creativity. These advancements are progressively rendering previously challenging biological pathways therapeutically accessible, paving the way for sophisticated precision medicine at an increasingly refined molecular level.
As boundaries between modalities continue to merge, we are entering an exciting new era where therapeutic strategies are guided by biological rationale rather than traditional, narrow molecular classifications, enabling targeted, effective, and personalised solutions for challenging targets and complex diseases.
The future of drug discovery promises unprecedented opportunities for innovation and impact—now is the time to harness this momentum.
Guest blog by Tom Fleming MChem MBA, CEO & Co-Founder, Arctoris
*If you would like to submit a guest blog, please email [email protected]